
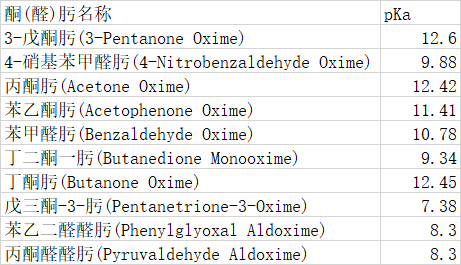
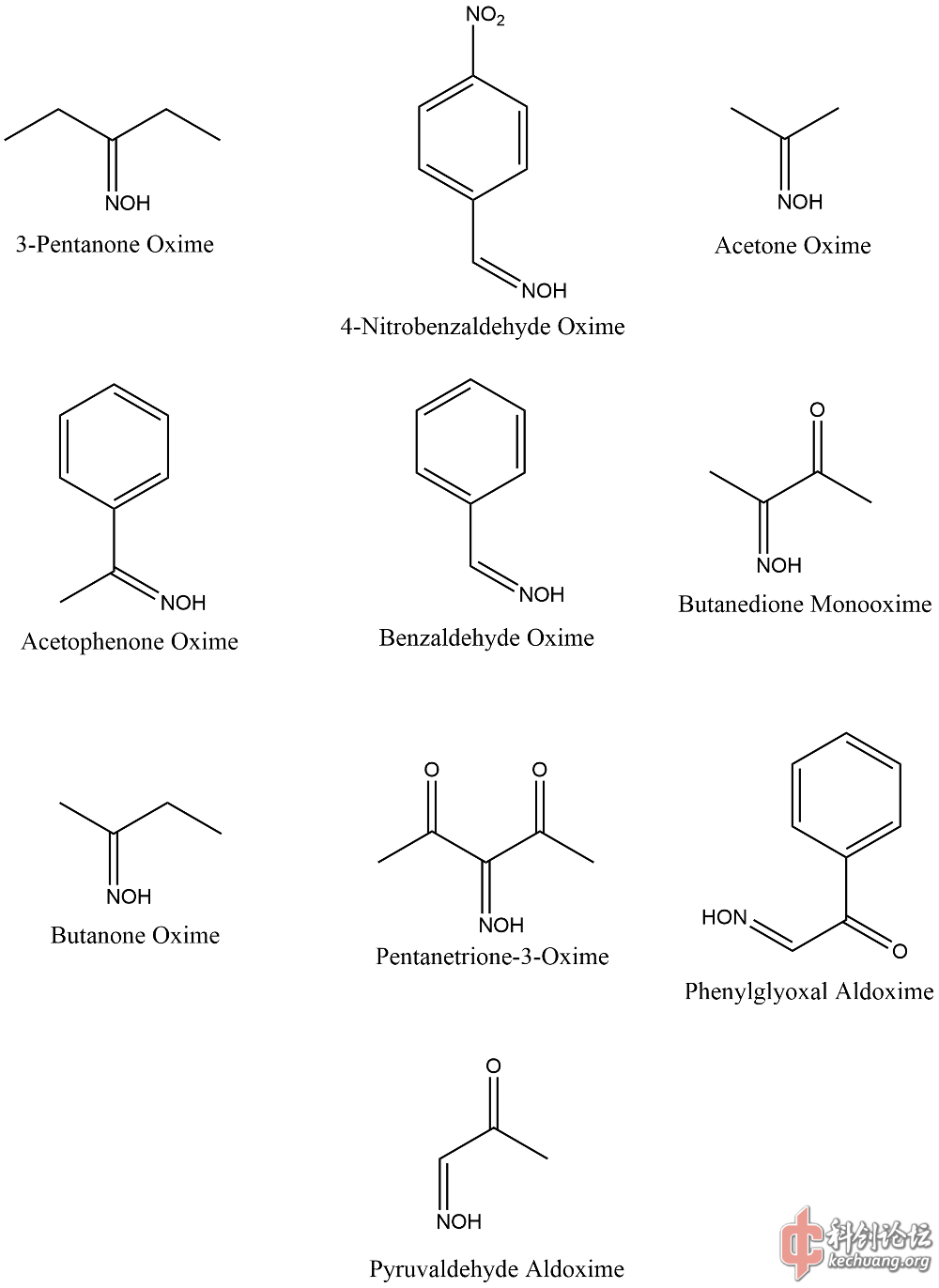














引用 last-order:半经验法我也觉得不准确,文献中也没有这么说,但毕竟需要实验数据,所以我也拿不准,本来就像看看大家的意见
首先鼓励楼主发帖,但有一些存在的问题我必须讲一讲。(原文献没有看(其实我手头也有一篇计算pKa的综述,不过我也没有看))
我个人觉得使用DFT方法计算应该算“从头算”,半经验这种说法感觉不太合适。
本质上来讲,计算pKa就是要得到电离过程的准确的Gibbs自由能变。因此应当选择足够准确的理论水平进行计算。
楼主使用的泛函B3LYP应该没太大问题(不过计算有机物热力学量更推荐使用M06-2X),但是基组的使用很不合适。6-31+G这个基组没有极化函数,很不可取。通常而言,基组中的极化函数是不能缺少的,而弥散函数则在几何优化及频率计算时可以不加。为了得到准确的热力学量,还应当在高理论水平下计算单点能(你的文中提到没有算氢离子是因为该物种无电子,所以没有电子能,热力学校正部分只存在平动能\\(3/2 RT\\))。
所以我建议的计算的理论水平是,在M062X/defSVP水平下优化和频率分析(使用相应的校正因子),在B2PLYP-D3/def2TZVP水平下计算单点能(如果计算资源吃紧,那就用M062X/def2TZVP)。
关于溶剂模型,目前比较推荐使用SMD模型。而IEFPCM比Gaussian默认的PCM模型还差,尽量别用吧23333
其实我想说的是,很多文献使用的计算方法并不一定合理。除非是一定要重复文献,自己动手算的时候可以自己考虑一下有没有更合理的理论方法。
前面提到的那个综述是doi: 10.1002/wcms.1218,有兴趣可以看一看。
关于纯理论计算,文献中也说过“The computational determination of accurate pKa values is very demanding, as an error of only 1.36 kcal/mol in ΔGaq is quivalent to a unit difference in pKa value at room temperature.”
基组选择时加极化了,你可能没看见。关于弥散,因为有阴离子,所以还是加了。基组不大,分子也不大,所以在最后一两次优化时加上弥散什么的计算量也能承受。
溶剂模型同时选择IEFPCM和SMD是在Gaussian的手册中看到的,说是更好
很感谢如此认真的回答和建议
200字以内,仅用于支线交流,主线讨论请采用回复功能。