

强烈支持这样的理论工作!
加载中
加载中
表情图片
评为精选
鼓励
加载中...
文件下载
加载中...
介绍:
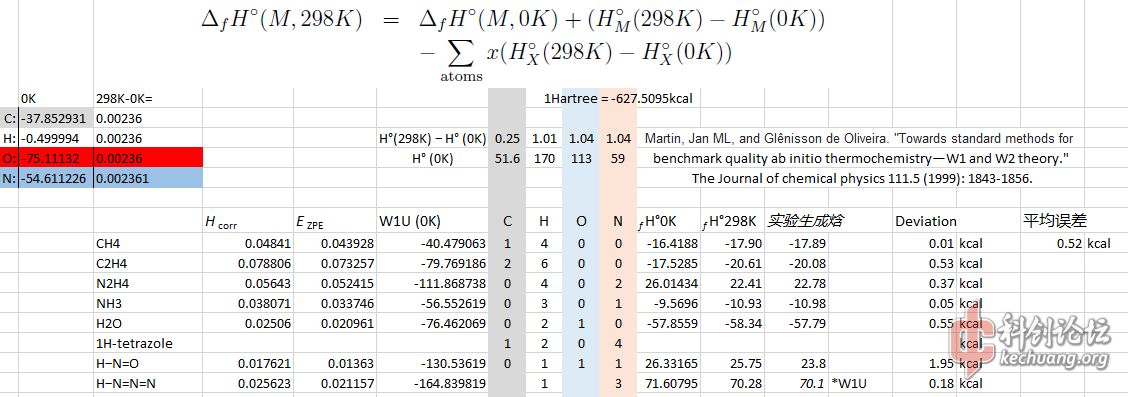
先感谢一下 dracula1429 在计算上的帮助。W1 类理论是计算化学里,气态生成焓计算精度最高的方法之一。W1U 的气态生成焓和实验结果相比,平均误是惊人的 0.3 kcal ,而却完全不带入经验法。
W1 使用个方法使用 coupled cluster (CC) 数值技术。CC 采用基本的Hartree-Fock 分子轨道方法,并使用指数聚类算子构造多电子波函数以解释电子相关性。该计算类似于CBS-X,基函数无限制外推。
W1U 的高精度也是由于使用了非常大的基组:高达cc-pVQZ + 2d1g和 cc-pV5Z + 2d1f 以及CCSD和CCSD(T)水平,所以如果计算材料超过2个重原子(碳,氧,氮,等等在W1U 里都算是重原子)的材料建议使用超级电脑或者高核心数量工作站。以下计算是使用Gaussian 09在中等配置私人电脑 ( 8 线程Xeon E3-1505M v5 带有 32GB RAM )得出的结果。
计算结果:
以下是7个小型材料的生成焓计算。其中,HN3,拥有3个重原子,和 C2H6 ,2个重原子,暂时是个人最大的计算材料,C2H6 耗时 4小时25 分钟。 1H-四唑跑了8个小时候出现了 diskspace 错误, 肯定是超越我电脑的极限了。
所有气态生成焓是在 NIST 数据库里找出。其中,HN3 的 70.1 气态生成焓不是实验数据,是和俄罗斯人使用超级计算机使用同样 W1U 方法得到的结果。2个结果,拥有 0.18 kcal 的误差,算比较吻合。
俄罗斯人的数据在这里:https://pubs.acs.org/doi/suppl/10.1021/jp404484q/suppl_file/jp404484q_si_001.pdf
W1 理论:https://aip.scitation.org/doi/abs/10.1063/1.479454
总结:
平均误差稍高于推广的 0.3 kcal 是因为 H-N=O 的气态生成焓和实验结果拥有 1.9kcal 的错误,个人认为这个可能是实验误差。
相对而言,该方法精度特别高。N2H4 的误差只有 0.37 kcal 的误差,在 B3LYP 6-31g(d) 的计算结果里,N2H4 的误差接近 10kcal,而是用同样方法带入到 1999 rice et al 方法后,误差更高。N2H4 在 B3LYP 6-311g+(2d,p) 精度的计算的结果和实验只有 2kcal 差距。
计算时间: W1> G2, CBS-APNO > G4 > G3, G2MP2 > CBS-QB3 > CBS-4M
精度:W1 > CBS-APNO > CBS-QB3, G4 > G3 > G2, G2MP2, CBS-4M
[修改于 6年9个月前 - 2018/08/22 12:30:13]
200字以内,仅用于支线交流,主线讨论请采用回复功能。