肟基(C=NOH)(之前错写为=NOH,经坛友指出后改正)许多人都比较陌生,但在医药领域和杂环合成方面都比较重要。肟基含有一个羟基,因此和醇一样,有一定酸性。由于氮的电负性较大,因此肟的酸性比醇大不少。有时需要在合成前了解它们的解离常数,因此较为准确简单的预测就显得比较重要。
分子动力学模拟极其耗时,因此理论计算就成了唯一可行的办法。既然这是一个平衡,那么就可以通过计算在水中电离时吉布斯自由能的变化来计算出平衡常数。
pKa = ΔGaq/(RT ln 10) ) = ΔGaq/(2.303RT)
然而实际并不是这么简单。从公式中就可以看出一点误差就会造成结果数量级的变化。肟电离出一个氢离子的能量变化当然容易计算,然而氢离子与大量水分子形成的N水合氢离子的过程,以及电离后结构的改变造成的水和能的变化等过程却难以精确计算。因此通过纯理论的方法在目前是几乎不可能的,于是就有了半经验法。某一类化合物因为基团相同,比较的话就会发现,解离常数只与其解离出氢离子的过程焓变有关,其它能量变化基本一致。因此很容易通过统计的方式得到解离出氢离子的能量变化与pKa的关系。
pKa = αΔH+β
文献中用的就是这种方法,不过更简单,连氢离子的能量都约掉了,用量子化学计算肟和肟根的能量差即可。
坛友补充:没有算氢离子是因为该物种无电子,所以没有电子能,热力学校正部分只存在平动能3/2RT
我用的是Gaussian09
方法选择:文献中说OLYP比B3LPY好,计算量小,结果更准确。我做了几个对比发现OLYP计算量确实小一些,但准确度没有什么优势。考虑到B3LYP较为常用,所以选择B3LYP。基组选择6-31g+ d。溶剂模型选择IEFPCM和smd。
样本选择
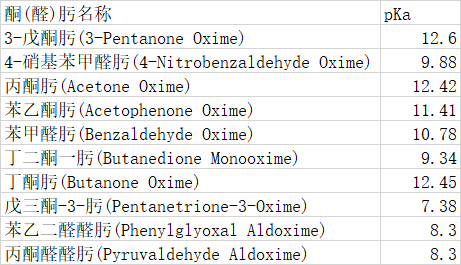
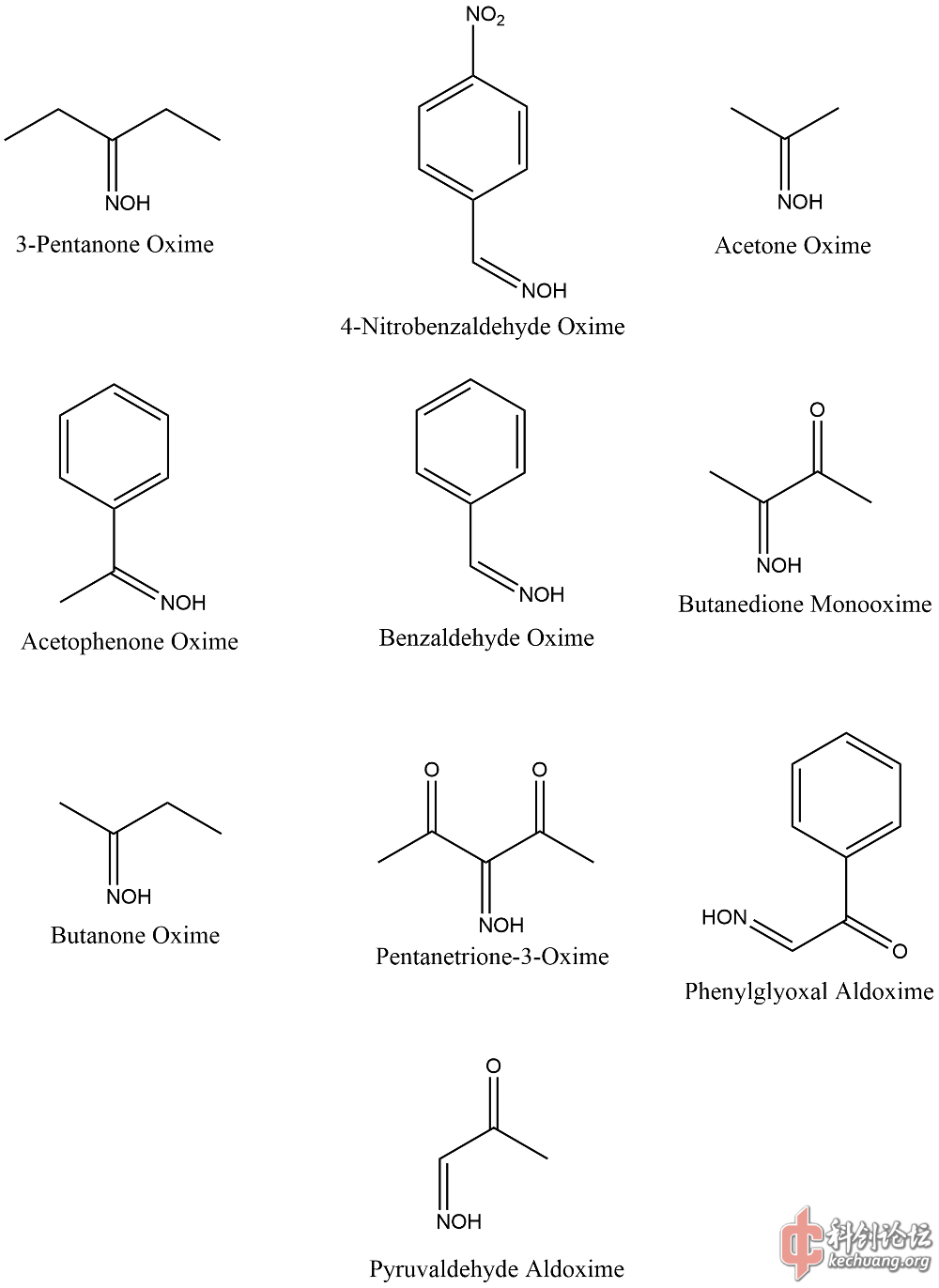
通过文献间对比,这些样本可信度较高。
第一步,分子的几何优化,下面是其能量最低构型
![3.png]()
![4.png]()
![5.png]()
![6.png]()
![7.png]()
![8.png]()
![9.png]()
![10.png]()
![11.png]()
第二步,计算它们的焓。
第三步,去掉一个氢并加上一个负电荷重复一二步
计算结果
![12.png]()
得出规律
![13.png]()
回归系数达到0.9868,线性关系较好
最后还要说明一下,此数据仅适合酮肟和醛肟,其他氯肟偕胺肟等并不适合
制备DNTF的第一步产物肟基丙二腈是一种酸性比较强的酮肟,那么酸性具体多大可以算一下看看
在刚才的条件下进行几何优化并计算焓
![14.png]()
焓为-354.252249 Ha
去掉一个氢并加上一个负电荷重复
![15.png]()
焓为-353.827782 Ha
计算pKa为 4.48
参考文献
 A Reliable and Efficient First Principles-Based Method for Predicting pKa Values.1.Methodology.pdf
741.77KB
PDF
367次下载
预览
A Reliable and Efficient First Principles-Based Method for Predicting pKa Values.1.Methodology.pdf
741.77KB
PDF
367次下载
预览
















200字以内,仅用于支线交流,主线讨论请采用回复功能。