


第一张图不够准确,谐振子模型实际上是一种简化了的分子震荡模型,目的是方便计算,二次函数的抛物线实际上是在描述震荡的大体范围,通常情况下,无论是写成概率波的p 还是写成震荡分布波,很难超越这个二次函数的外界,所以你那张图是不够准确的,这实际上就是求一个H普赛=E普赛的问题,求二阶偏微分然后带回去让他们相等。在较复杂的计算中,也会用到变分函数,目的是试探出待定系数。
81454
%7B%22isLastPage%22%3Atrue%2C%22notes%22%3A%5B%5D%2C%22pid%22%3A%22831378%22%2C%22tid%22%3A%2281454%22%2C%22mainForumsId%22%3A%5B%2283%22%5D%2C%22categoriesId%22%3A%5B%220%22%5D%2C%22tcId%22%3A%5B%5D%7D
%7B%22isEditMode%22%3Afalse%7D
Numerov方法数值求解一维量子谐振子波函数
现在在自学赖文的量子化学,最近看到简谐振子这一部分。书中以简谐振子为例,介绍了使用Numerov方法数值求解Schroedinger方程的方法,并提供了一个C++的代码来实现。因为我不会C++,所以就使用正在学的Python来尝试写一个(刚学到函数)。
原理
我在维基上搜了一下,Numerov方法不只用于量子力学。但是涉及的其他的领域我不了解,所以无法介绍。总之就是单纯的讲讲求解一维谐振子波函数的方法。
$$ \hat{H}=\hat{T}+\hat{V}=-\frac{\hbar^2}{2m}\ \frac{d^2}{dx^2}+\frac{1}{2}\ kx^2=-\frac{\hbar^2}{2m}\ \frac{d^2}{dx^2}+2\pi^2\nu^2mx^2=-\frac{\hbar^2}{2m}\ (\frac{d^2}{dx^2}-\alpha^2x^2) $$ $$ \alpha=\frac{2\pi\nu m}{\hbar} $$
一维量子谐振子的Schroedinger方程: $$ \frac{d^2\psi}{dx^2}+(2mE\hbar^{-2}-\alpha^2 x^2)\psi=0 $$
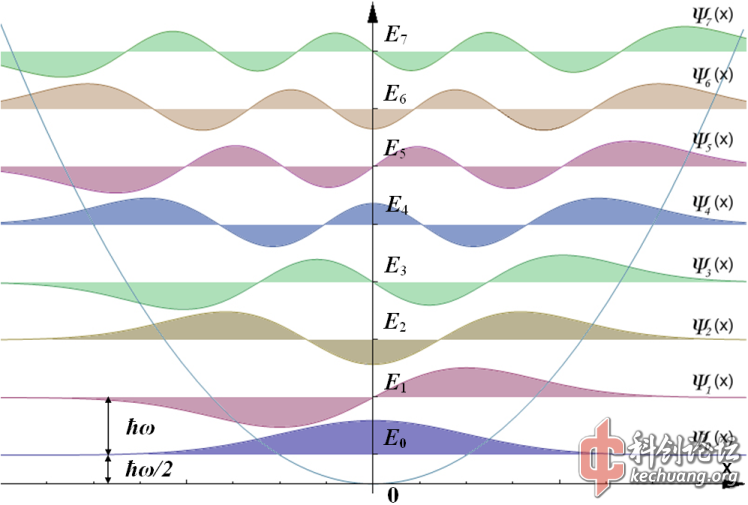
一维谐振子的解析解的求解不算很难,这里也不做介绍了。
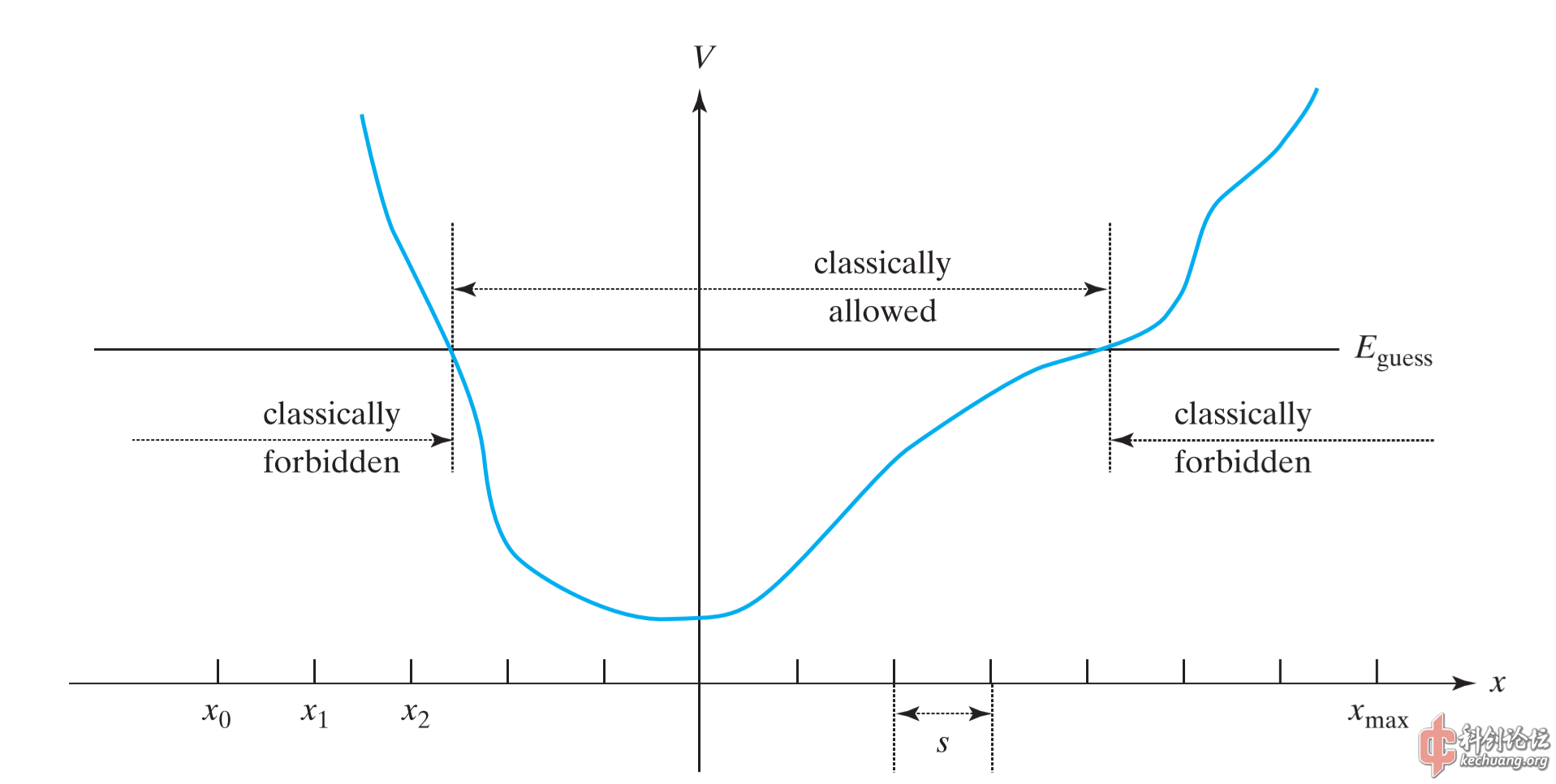
Numerov方法是基于递推,将我们所探讨的区域划分为多个小的间隔\(s\)。若我们取最左端的点的坐标为\(x_0\),向右的点分别为\(x_1=x_0 +s,x_2=x_1 +s, \ldots ,x_\text{max} \)。设\(\psi_n, \psi_{n+1}, \psi_{n-1}\)分别表示\(\psi\)在\(x_n, x_n+s, x_n-s\)处的值:
$$ \psi(x_n)=\psi_n, \psi(x_n+s)=\psi_{n+1}, \psi(x_n-s)=\psi_{n-1} $$
将Schroedinger方程写为:
$$ \psi''=G\psi, G=m\hbar^{-2}[2V(x)-E] $$
把\(\psi(x_n+s)\)和\(\psi(x_n-s)\)进行Taylor展开:
$$ \psi(x_n+s)=\psi(n)+\psi'(n)\cdot s+\frac{\psi''(n)}{2}\cdot s^2+\frac{\psi'''(n)}{3!}\cdot s^3+\frac{\psi^\text{(4)}(n)}{4!}\cdot s^4+\frac{\psi^\text{(5)}(n)}{5!}\cdot s^5+\ldots $$
$$ \psi(x_n-s)=\psi(n)-\psi'(n)\cdot s+\frac{\psi''(n)}{2}\cdot s^2-\frac{\psi'''(n)}{3!}\cdot s^3+\frac{\psi^\text{(4)}(n)}{4!}\cdot s^4-\frac{\psi^\text{(5)}(n)}{5!}\cdot s^5+\ldots $$
将两式相加(忽略后面的项):
$$ \psi_{n+1}=-\psi_{n-1}+2\psi_n+\psi''_n\cdot s^2+\frac{\psi^\text{(4)}_n}{12}\cdot s^2 $$
用\(\psi''\)代替\(\psi\),并两边乘以\(s^2\):
$$ \psi''_{n+1} \cdot s^2=-\psi''_{n-1}\cdot s^2+2\psi''_n\cdot s^2+\psi^\text{(4)}_n\cdot s^4+\frac{\psi^\text{(6)}_n}{12}\cdot s^6 $$
忽略含6阶导的项:
$$ \psi^\text{(4)}_n\cdot s^4=\psi''_{n+1}\cdot s^2+\psi''_{n-1}\cdot s^2-2\psi''_n\cdot s^2 $$
将上式代入之前的式子,并代入\(\psi''=G\psi\):
$$ \psi_{n+1}=-\psi_{n-1}+2\psi_n+G_n\psi_n s^2+\frac{1}{12}\ [G_{n+1}\psi_{n+1}s^2+G_{n-1}\psi_{n-1}s^2-2G_n\psi_n s^2] $$
所以:
$$ \psi_{n+1}=\frac{2\psi_n-\psi_{n-1}+5G_n\psi_n s^2 /6+G_{n-1}\psi_{n-1}s^2 /12}{1-G_{n+1}s^2 /12} $$ $$ G_n=G(x_n)=m\hbar^{-2}[2V(x_n)-2E] $$
由此我们得到了该递推关系式。为了用该式求得波函数,我们首先需要得到\(\psi_0, \psi_1\)。 根据经典力学,\(E<V\)的区域为禁阻区域;而在量子力学中,由于隧穿效应,在\(E<V\)的区域,波函数\(\psi\)并不为零。但是进入经典禁阻区域越深,\(\psi\)越趋于零。因此我们假定\(\psi(x_0)=\psi_0=0\)。而对于\(x_1\)点,假设波函数在该点为一很小的值,比如可以取0.0001。事实上取0.001也是可以的,这相当于将波函数乘以10,通过归一化处理后并没有什么区别。
要进行递推,显然我们还需要猜测一个本征能量值\(E_\text{guess}\)。经过递推最终得到\(\psi(x_\text{max})\)。根据品优波函数平方可积或对称性的要求,\(\psi(x_\text{max})\)应当趋近于零。若\(\psi(x_\text{max})\)远远偏离0,说明我们猜测的能量本征值不够合理,需要重新进行猜测。
有了上述的方法,我们尝试用程序进行实现。
程序实现
为了方便地进行计算,首先我们要做的一件事是将前面涉及的式子去量纲化(更准确的说法是将其转换为自然单位制)。
具体的变换不写了,并不算难。最终变换得到的Schroedinger方程为: $$ \frac{d^2\psi_r}{dx^2_r}=(2V_r-2E_r)\psi_r $$
$$ \psi''_r=G_r \psi_r, G_r=2V_r-2E_r $$
上式中:\(V_r=\frac{1}{2}\ x_r^2\)。
经过处理,这些式子中的各个量的量纲均为1。
然后就是代码实现了。
(瞎画了一个流程图)

然后是python代码:
<code class="language-python">X={"x0":0.0}
P={"p0":0.0}
G={"g0":0.0}
nn=0
x=float(input("Enter initial xr:"))
s=float(input("Enter the increment sr:"))
m=int(input("Enter the number of intervals m:"))
E=float(input("Enter the reduced energy Er:"))
for i in list(range(m+1)):
X["x%s" % i]=0.0 #储存x
for i in list(range(m+1)):
P["p%s" % i]=0.0 #储存psi
for i in list(range(m+1)):
G["g%s" % i]=0.0 #储存g
P["p1"]=0.0001
X["x0"]=x
X["x1"]=X["x0"]+s
G["g0"]=X["x0"]*X["x0"]-2*E
G["g1"]=X["x1"]*X["x1"]-2*E
ss=s*s/12
for i in range(1,m+1):
X["x%s" % (i+1)]=X["x%s" % i]+s
G["g%s" % (i+1)]=X["x%s" % (i+1)]*X["x%s" % (i+1)]-2*E
P["p%s" % (i+1)]=(-P["p%s" % (i-1)]+2*P["p%s" % i]+10*G["g%s" % i]*P["p%s" % i]*ss+G["g%s" % (i-1)]*P["p%s" % (i-1)]*ss)/(1-G["g%s" % (i+1)]*ss)
if (P["p%s" % i]*P["p%s" % (i+1)]<0): nn="nn+1" print("er="+str(E))
print(" nodes="+str(nn))
for i in range(m+1):
print(" psir(%f)="%f"" % (x["x%s" i],p["p%s" i])) < code></0):></code>
这个代码实际上写的很拙劣,因为我目前只学了一点,各位不要嘲笑……
关于输入的一点说明:为了让得到的结果尽可能精确,\(s\)的选择应当尽量小一些,这样可以绘制更多的点。m为间隔的数量,显然\(m=\frac{(x_\text{max}-x_0)}{s}\)。
运行该代码,写入输入,则得到输出(同时会计算节点的数量)。
<code class="language-python">Enter initial xr:-4 Enter the increment sr:0.05 Enter the number of intervals m:160 Enter the reduced energy Er:0.5 Er=0.5 Nodes=0 Psir(-4.000000)=0.000000 Psir(-3.950000)=0.000100 Psir(-3.900000)=0.000204 Psir(-3.850000)=0.000315 ... </code>
其中最后一行为\(x_\text{max}\)的波函数值:
<code class="language-python">Psir(4.000000)=0.000489 </code>
可以看出,与0很接近。
若其偏离0,则需要手动对能量进行修改再次进行计算。
用以上数据在Origin中绘图:

与量子谐振子基态的波函数非常吻合(输出中表明了节点数为0)。
若修改初猜能量,得到第一激发态的波函数也是同样可行的。
但需要注意的一点是,当振动量子数为奇数时,\(\psi\)首先是小于零的。因此需要修改代码中的P["p1"]=0.0001为-0.0001。
P.S.书中提供了一段C++实现代码:
修改版代码
实际使用代码尝试后会发现\(E_0, E_1, E_2,\ldots\)分别近似等于0.5,1.5,2.5...(不是严格相等大概是程序计算时数值精度的原因)
这是非常理所当然的一点,因为量子谐振子的能级满足:
$$ E_v=(v+\frac{1}{2}\ )h\nu $$
约化后能量自然等于0.5,1.5,2.5...
(以下内容我也不知道算不算画蛇添足)
所以我考虑通过修改代码,通过输入振动量子数,再利用程序自动对能量进行调整(由于程序原因,输入精确本征值,最后一点的波函数仍不够趋于0),最后进行数值计算。
所以我在进行最终计算前,设定一收敛条件,若\(\psi(x_\text{max})\)的绝对值小于0.0001,则判断\(E_\text{guess}\)足够准确。并由此进行计算。
修改后的代码如下(同求不嘲笑……):
<code class="language-python">X={"x0":0.0}
P={"p0":0.0}
G={"g0":0.0}
nn=0
x=float(input("Enter initial xr:"))
s=float(input("Enter the increment sr:"))
m=int(input("Enter the number of intervals m:"))
v=float(input("Enter the vibration quantum number:"))
if v%2==0:
P["p1"]=0.0001
else:
P["p1"]=-0.0001
p1=P["p1"]
E=v+0.5
El=v
Eh=v+1
M=E
#定义函数求最后一点的psir
def iteration(E,x0,s,m,p1):
p0=0.0
x1=x0+s
x2=x1+s
ss=s*s/12
for i in range(m-1):
g0=x0*x0-2*E
g1=x1+x1-2*E
g2=x2*x2-2*E
p2=(-p0+2*p1+10*g1*p1*ss+g0*p0*ss)/(1-g2*ss)
x0=x0+s
x1=x1+s
x2=x2+s
p0=p1
p1=p2
m=m-1
return p2
#二分法
while abs(iteration(E,x,s,m,p1))>0.0001:
if iteration(E,x,s,m,p1)>0:
if iteration(Eh,x,s,m,p1)<0: e="(E+Eh)/2" if iteration(e,x,s,m,p1)>0:
pass
else:
Eh=E
E=M
else:
E=(E+El)/2
if iteration(E,x,s,m,p1)>0:
pass
else:
El=E
E=M
else:
if iteration(Eh,x,s,m,p1)<0: e="(E+El)/2" if iteration(e,x,s,m,p1)<0: pass else: el="E" eh="E" x["x0"]="x" x["x1"]="X["x0"]+s" g["g0"]="X["x0"]*X["x0"]-2*E" g["g1"]="X["x1"]*X["x1"]-2*E" ss="s*s/12" for i in range(1,m): x["x%s" % (i+1)]="X["x%s"" i]+s g["g%s" (i+1)]*x["x%s" (i+1)]-2*e p["p%s" (i-1)]+2*p["p%s" i]+10*g["g%s" i]*p["p%s" i]*ss+g["g%s" (i-1)]*p["p%s" (i-1)]*ss) (1-g["g%s" (i+1)]*ss) (p["p%s" (i+1)]<0): nn="nn+1" print("er="+str(E))
print(" nodes="+str(nn))
for i in range(m+1):
print(" psir(%f)="%f"" (x["x%s" i],p["p%s" i])) < code></0:></0:></code>
在上面的代码中,我定义了一个函数用于求\(\psi(x_\text{max})\),再利用二分法调整能量\(E\),当\(\psi(x_\text{max})\)满足收敛条件后,便使用调整后的能量计算各点波函数的数值(这部分与第一段代码没有区别)。
从原理上来说,这段代码应该没有明显的毛病。但是发现得到的能量并不准确。
测试后发现是因为函数的输出与第一段代码的输出有明显的差距。
把函数的代码单独运行,并使其输出每一次得到的\(\psi\)(前两行是我手动补上去的)
<code class="language-python">0.0000000000 0.0001000000 0.00019873416928030692 0.00029497854356514764 0.00038754990743566356 0.00047532063060121913 0.0005572325408706247 0.0006323096548955608 0.0006996696081080572 ... 0.26353345349798035 0.3024049078455913 0.3474895062785628 </code>
这是第一段代码的输出:
<code class="language-python">Psir(-4.000000)=0.000000 Psir(-3.950000)=0.000100 Psir(-3.900000)=0.000204 Psir(-3.850000)=0.000315 Psir(-3.800000)=0.000436 Psir(-3.750000)=0.000573 Psir(-3.700000)=0.000728 ... Psir(3.900000)=0.000542 Psir(3.950000)=0.000506 Psir(4.000000)=0.000489 </code>
可以看出两段代码的输出结果的相差程度越来越大,到了最后一行,结果已经相差近三个数量级。用该函数的结果来调整能量的优化自然难以得到准确的结果。
我仔细检查了函数的代码,自认为没有错误。所以怀疑是浮点数精度(计算和储存时的四舍五入)的原因。
所以目前第二段代码暂不用来计算。
[修改于 7年10个月前 - 2017/03/02 13:13:37]

引用 QQQQQQQ:
第一张图不够准确,谐振子模型实际上是一种简化了的分子震荡模型,目的是方便计算,二次函数的抛物线实际上是在描述震荡的大体范围,通常情况下,无论是写成概率波的p 还是写成震荡分布波,很难超越这个二次函数……
简谐振子确实是一个非常理想化的模型,实际双原子分子的振动也偏离简谐振动。
其实我是考虑过以后将势能项改为Morse势能,再尝试数值求解的问题。
至于变分法,那更是要等以后才能have a try233333
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。

赞。
大致浏览了一下,这是一个用数值试探解法解本征振动模式的方法,思路相当直观(对于我这样理解不了高级方法的,也很喜欢这样的方法)。
在费曼物理学讲义第三册16章6节,“量子化能级”,提示过类似的方法。那本书写成于1960年代早期,计算机数值方法刚刚起步。
基本思路是把待求的本征模波函数(量子力学管它叫定态波函数)分解成两个函数相乘,这里假设为A和B相乘。A只以时间坐标为自变量,B只以空间坐标为自变量。为什么可以这么分呢?因为本征振动模式的特点是随时间简谐变化,可以如此分解。
然后时间部分A自然按照普朗克关系E=h*f按照时间简谐推演。
空间部分按照德布罗意关系P=h*k按照空间简谐推演。
E和P的关系由粒子能量和势阱函数的关系得到。对于经典情形就是 E总=E动+E势。动能E动和动量P由经典力学关系 E动=P^2/2m 关联。
有了这些关系,即可在空间中从边界开始逐步推演波函数B了。注意要点是k随空间的势能而逐点改变。
推到另外的边界后,观察收敛和对称情况。根据观察到的信息,修改初始条件重新推演,直到得到满足条件的解。这些解可以有多个,对应多个本征模式。
把这里的物理参数改换一下,可以应用于很多其它物理问题。比如说,我过去研究多模光纤的传输模时,曾经编过一个原理基本相同的matlab程序,可以精确地导出多模光纤的所有传输模。令人惊叹的是,如此简单的方法,得到的结果精度居然超过我旁边一博士用三维电磁建模程序得到的结果。
大致浏览了一下,这是一个用数值试探解法解本征振动模式的方法,思路相当直观(对于我这样理解不了高级方法的,也很喜欢这样的方法)。
在费曼物理学讲义第三册16章6节,“量子化能级”,提示过类似的方法。那本书写成于1960年代早期,计算机数值方法刚刚起步。
基本思路是把待求的本征模波函数(量子力学管它叫定态波函数)分解成两个函数相乘,这里假设为A和B相乘。A只以时间坐标为自变量,B只以空间坐标为自变量。为什么可以这么分呢?因为本征振动模式的特点是随时间简谐变化,可以如此分解。
然后时间部分A自然按照普朗克关系E=h*f按照时间简谐推演。
空间部分按照德布罗意关系P=h*k按照空间简谐推演。
E和P的关系由粒子能量和势阱函数的关系得到。对于经典情形就是 E总=E动+E势。动能E动和动量P由经典力学关系 E动=P^2/2m 关联。
有了这些关系,即可在空间中从边界开始逐步推演波函数B了。注意要点是k随空间的势能而逐点改变。
推到另外的边界后,观察收敛和对称情况。根据观察到的信息,修改初始条件重新推演,直到得到满足条件的解。这些解可以有多个,对应多个本征模式。
把这里的物理参数改换一下,可以应用于很多其它物理问题。比如说,我过去研究多模光纤的传输模时,曾经编过一个原理基本相同的matlab程序,可以精确地导出多模光纤的所有传输模。令人惊叹的是,如此简单的方法,得到的结果精度居然超过我旁边一博士用三维电磁建模程序得到的结果。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。

Numerov方法本身是一个解常微分方程的线性多步法,只要是缺一阶项的二阶常微分方程就可用。
看来这个算法的想法是猜出一个能量本征值,再用其尝试数值求解波函数,判断与边界条件的符合程度再调整猜测的能量本征值,是一种处理本征值问题的常见手段。
但是其局限性也很大,这样的方法只能方便的处理一维问题,超出最简单的这些toy model就无能为力了,据我所知在真正的计算化学中也不重要。
看来这个算法的想法是猜出一个能量本征值,再用其尝试数值求解波函数,判断与边界条件的符合程度再调整猜测的能量本征值,是一种处理本征值问题的常见手段。
但是其局限性也很大,这样的方法只能方便的处理一维问题,超出最简单的这些toy model就无能为力了,据我所知在真正的计算化学中也不重要。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用 qiuzheru:说得没错。(就连处理的问题本身都属于最简单的模型之一)
Numerov方法本身是一个解常微分方程的线性多步法,只要是缺一阶项的二阶常微分方程就可用。
看来这个算法的想法是猜出一个能量本征值,再用其尝试数值求解波函数,判断与边界条件的符合程度再调整猜测的能量……
前几天把代码稍微修改了一下,也可以用于数值求解氢原子径向波函数。
但对我来说,这些都是toy problem,就是想练习一下写代码(虽说也没有太复杂)。
如前面所说的,我在一段时间内的目标是写出一个能够求解HF的程序。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。