80908
%7B%22isLastPage%22%3Atrue%2C%22notes%22%3A%5B%5D%2C%22pid%22%3A%22826980%22%2C%22tid%22%3A%2280908%22%2C%22mainForumsId%22%3A%5B%2283%22%5D%2C%22categoriesId%22%3A%5B%22336%22%5D%2C%22tcId%22%3A%5B%5D%7D
%7B%22isEditMode%22%3Afalse%7D
量子化学软件计算:含能离子晶体密度的预测

浏览本文前请先阅读以下两篇文章,可以获得更好体验。
量子化学软件计算:以三肼基均三嗪的密度、生成焓为例
XXXXXXXXXXXXXXXXXXXXXXXX/t/80709
量子化学软件计算:含CHNO中性分子的密度预测
XXXXXXXXXXXXXXXXXXXXXXXX/t/80801
本文仍然使用ChemBioOffice 2014、Gaussian 03w、Multiwfn等软件,并按照Politzer拟合针对含能离子晶体的参数和公式对软件计算结果进行处理。
在之前的帖子中已详细展现了软件的使用及数据的处理、读取,本文即不再累述重复部分,仅将不同于中性体系的内容加以介绍。
一、Politzer参数和公式
Politzer拟合得到的公式为:
density = αM/Vm + β(Vs+/As+) + γ(Vs-/As-) + δ(1)
其中:α = 1.0260
β = 0.0514
γ = 0.0419
δ = 0.0227
M = 相对分子质量
Vm = 分子体积
Vs+ = Positive average value(阳离子表面为正的区域平均静电势)
As+ = Positive surface area(阳离子表面为正的区域面积)
Vs- = Negative average value(阴离子表面为负的区域平均静电势)
As- = Negative surface area(阴离子表面为负的区域面积)
二、举例示范
以高氯酸铵为例对软件及操作进行示范举例。
首先在Chem 3D中分别绘制出NH4+离子和ClO4-离子,形成两个文件,将NH4+离子和ClO4-离子分开处理得到各自独立的数据。如图2.1。
![1.jpg]()
![2.jpg]()
图2.1 独立的NH4+离子和ClO4-离子
然后使用Chem 3D内嵌的Gaussian对过渡态进行优化,得到fchk文件后再使用Multiwfn打开fchk文件,处理得到离子数据,如图2.2。
![3.jpg]()
图2.2 ClO4-离子的各项参数
将得到的NH4+离子和ClO4-离子参数代入式1,各项参数如表2.1。
表2.1 得到的NH4ClO4各项参数
![4.jpg]()
其中Vm为NH4+离子和ClO4-离子体积之和。注意在计算二盐类时Vm为阳离子与两个阴离子体积之和。
三、精度
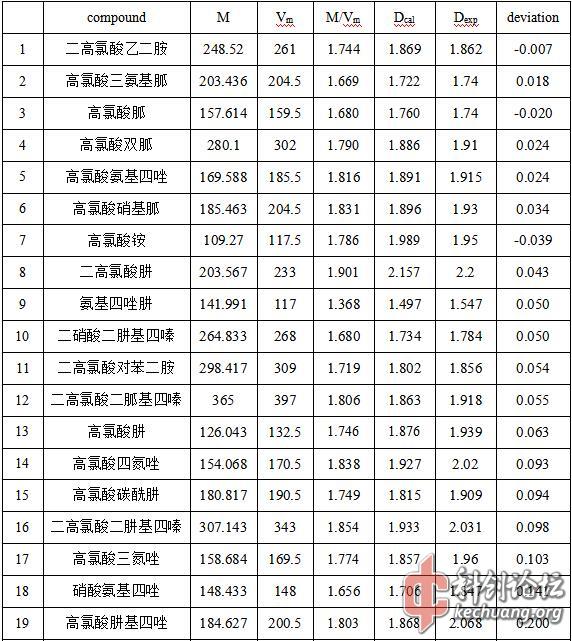
大量浏览国内外文献可查,离子晶体的密度预测往往偏差较大。而本文采用的方法经验证在预测阳离子呈链状有机碱时(脂肪胺类、胍类、肼类)其准确度较高,误差往往不会超过0.05 g/cm3;氮杂环有机碱类离子晶体密度预测偏低,误差在0.1~0.2 g/cm3;介于二者之间的连接有较大链状基团的氮杂环有机碱误差小于氮杂环有机碱,大于链状有机碱;芳香胺类误差较脂肪胺类增大不明显;含羰基类有机碱误差较不含羰基的大。如表3.1。
表3.1 误差统计
![5.jpg]()
四、结论
1、本文采用的方法得到的离子数据与Politzer发表的文献内给出的高度相符,验证了本文所采用的软件及计算方法的正确性。
2、本文方法预测阳离子呈链状有机碱时(脂肪胺类、胍类、肼类)其准确度较高,可有效指导新型含能材料设计工作;氮杂环有机碱类离子晶体密度预测偏低,采用本文方法时,注意纠正误差。
3、因统计样本较少,加上文献给出的部分样本密度不统一,存在误差。本文对连接有较大链状基团的氮杂环有机碱、芳香胺、含羰基有机碱的预测精度推断并不严谨,缺乏大量数据支持。
大量选取不同类型有机碱统计预测密度误差,可以有效统计出本文方法对不同类型有机碱的预测准确性,甚至可进一步针对不同类型修正参数,完善公式。
参考文献:Politzer et al., Mol. Phys., 108, 1391 (2010)
XXXXXXXXXXXXXXXXXXX/337
量子化学软件计算:以三肼基均三嗪的密度、生成焓为例
XXXXXXXXXXXXXXXXXXXXXXXX/t/80709
量子化学软件计算:含CHNO中性分子的密度预测
XXXXXXXXXXXXXXXXXXXXXXXX/t/80801
本文仍然使用ChemBioOffice 2014、Gaussian 03w、Multiwfn等软件,并按照Politzer拟合针对含能离子晶体的参数和公式对软件计算结果进行处理。
在之前的帖子中已详细展现了软件的使用及数据的处理、读取,本文即不再累述重复部分,仅将不同于中性体系的内容加以介绍。
一、Politzer参数和公式
Politzer拟合得到的公式为:
density = αM/Vm + β(Vs+/As+) + γ(Vs-/As-) + δ(1)
其中:α = 1.0260
β = 0.0514
γ = 0.0419
δ = 0.0227
M = 相对分子质量
Vm = 分子体积
Vs+ = Positive average value(阳离子表面为正的区域平均静电势)
As+ = Positive surface area(阳离子表面为正的区域面积)
Vs- = Negative average value(阴离子表面为负的区域平均静电势)
As- = Negative surface area(阴离子表面为负的区域面积)
二、举例示范
以高氯酸铵为例对软件及操作进行示范举例。
首先在Chem 3D中分别绘制出NH4+离子和ClO4-离子,形成两个文件,将NH4+离子和ClO4-离子分开处理得到各自独立的数据。如图2.1。

图2.1 独立的NH4+离子和ClO4-离子
然后使用Chem 3D内嵌的Gaussian对过渡态进行优化,得到fchk文件后再使用Multiwfn打开fchk文件,处理得到离子数据,如图2.2。

图2.2 ClO4-离子的各项参数
将得到的NH4+离子和ClO4-离子参数代入式1,各项参数如表2.1。
表2.1 得到的NH4ClO4各项参数

其中Vm为NH4+离子和ClO4-离子体积之和。注意在计算二盐类时Vm为阳离子与两个阴离子体积之和。
三、精度
大量浏览国内外文献可查,离子晶体的密度预测往往偏差较大。而本文采用的方法经验证在预测阳离子呈链状有机碱时(脂肪胺类、胍类、肼类)其准确度较高,误差往往不会超过0.05 g/cm3;氮杂环有机碱类离子晶体密度预测偏低,误差在0.1~0.2 g/cm3;介于二者之间的连接有较大链状基团的氮杂环有机碱误差小于氮杂环有机碱,大于链状有机碱;芳香胺类误差较脂肪胺类增大不明显;含羰基类有机碱误差较不含羰基的大。如表3.1。
表3.1 误差统计
四、结论
1、本文采用的方法得到的离子数据与Politzer发表的文献内给出的高度相符,验证了本文所采用的软件及计算方法的正确性。
2、本文方法预测阳离子呈链状有机碱时(脂肪胺类、胍类、肼类)其准确度较高,可有效指导新型含能材料设计工作;氮杂环有机碱类离子晶体密度预测偏低,采用本文方法时,注意纠正误差。
3、因统计样本较少,加上文献给出的部分样本密度不统一,存在误差。本文对连接有较大链状基团的氮杂环有机碱、芳香胺、含羰基有机碱的预测精度推断并不严谨,缺乏大量数据支持。
大量选取不同类型有机碱统计预测密度误差,可以有效统计出本文方法对不同类型有机碱的预测准确性,甚至可进一步针对不同类型修正参数,完善公式。
参考文献:Politzer et al., Mol. Phys., 108, 1391 (2010)
XXXXXXXXXXXXXXXXXXX/337
[修改于 8年2个月前 - 2016/10/20 22:29:08]

dracula1429
 专家 学者 机友 笔友
专家 学者 机友 笔友