65017
%7B%22isLastPage%22%3Atrue%2C%22notes%22%3A%5B%5D%2C%22pid%22%3A%22t65017%22%2C%22tid%22%3A%2265017%22%2C%22mainForumsId%22%3A%5B%2283%22%5D%2C%22categoriesId%22%3A%5B%22336%22%5D%2C%22tcId%22%3A%5B%5D%7D
%7B%22isEditMode%22%3Afalse%7D
调查:有多少人需要Gaussian09W+GaussView5.0量子化学计算软件?

本帖最后由 718281828kc 于 2014-4-28 22:32 编辑
Gaussian是一个功能强大的量子化学综合软件包。其可执行程序可在不同型号的大型计算机,超级计算机,工作站和个人计算机上运行,并相应有不同的版本。高斯功能:过渡态能量和结构、键和反应能量、分子轨道、原子电荷和电势、振动频率、红外和拉曼光谱、核磁性质、极化率和超极化率、热力学性质、反应路径,计算可以对体系的基态或激发态执行。可以预测周期体系的能量,结构和分子轨道。因此,Gaussian可以作为功能强大的工具,用于研究许多化学领域的课题,例如取代基的影响,化学反应机理,势能曲面和激发能等等。
Gaussian 09增加的功能
能量和求导
⑴最近发展的半经验模型(AM1,PM3,PM3MM,PDDG,PM6),计算解析一阶导和二阶导,用户自定义参数,以及结合使用PCM溶剂模型。
⑵TDDFT解析梯度和数值频率。
⑶EOM-CCSD计算激发能。
⑷新的DFT泛函,包括HSE,wB97,m05/m06,LC类泛函,以及双杂化B2PLYP。
⑸经验离散模型和相应的泛函。
⑹ROMP3,ROMP4,ROCCSD,ROCCSD(T)能量。
⑺W1RO,W1BD,G4方法计算能量。
⑻DFTB半经验模型,以及使用解析矩阵元的DFTBA版本。
ONIOM
⑴ONIOM与PCM组合。有多种ONIOM+PCM模型。
⑵ONIOM计算IRC,即使分子包含上千个原子效率也很高。
溶剂化
⑴新的PCM溶剂化算法,使能量成为核坐标的连续函数。现在,PCM的几何优化与气相优化的收敛速度一样。
⑵特定态的自洽溶剂化,用于模拟荧光和其它发射过程。它对上百种溶剂进行了参数化,可以给出非常好的总溶剂化自由能。
几何优化和IRC
⑴能量最小化默认使用GEDⅡS几何优化算法,这对大的柔软分子特别有帮助。
⑵对最小值和过渡结构使用二次收敛ONIOM(MO:MM)优化,既用于力学部分,也用于电子嵌入部分。
⑶一个输入部分,用于控制优化中的冻结或非冻结原子。可以用原子、元素、残基、或ONIOM层来指定原子。
分子特性
⑴解析的含频ROA强度。
⑵解析的DFT 超极化率。
⑶使用两个态的谐振模式,通过Franck-Condon原理计算电子激发、发射、光电离的谱带带型。
⑷用Herzberg-Teller或Franck-Condon-Herzberg-Teller理论计算电子激发的谱带带型。
⑸选择简正模式用于显示,非谐校正,和FC/HT/FCHT分析。可通过原子、元素、残基、或ONIOM层来选择。
分析和输出
⑴蛋白质二级结构的信息可以包含在分子指定输入部分,或者.fchk文件中。
⑵轨道布居分析,给出原子或角动量对轨道的贡献。
⑶正则UHF/UDFT进行二次正交化,用于显示或用于ROHF计算的初始猜测。
⑷CIS和TD激发的自然跃迁轨道分析。
⑸把占据轨道投影到最小基之后,进行Mulliken布居分析。当基组增大时,这能给出稳定的布居。
其它新功能
⑴SCF的初始猜测可以从片段计算的组合产生,需要指定每个片段的电荷和自旋。
⑵用四点差分而不是默认的两点差分计算数值频率,具有更高的精度和数值稳定性。
效率改善
⑴HF和DFT对大分子的频率计算更快,特别是当并行时。
⑵FMM以及线性标度的库仑和交换对簇并行。
⑶大体系的ONIOM(MO:MM)频率计算更快,特别是对电子嵌入。可以计算100-200 QM原子和6000 MM原子的频率。
⑷在大型频率计算中保存简正模式,用于显示或打印模式,以及开始IRC=RCFC任务。
⑸CC,BD,和EOM-CCSD振幅可以保存在检查点文件中,在以后的计算中读入。保存BD轨道并在以后读入。
⑹半经验,HF,和DFT的频率计算可以在中期计算中重新开始。
⑺CC和EOM-CC计算可以在中期计算中重新开始。
⑻ONIOM各步的初始猜测可以来自不同的检查点文件。
⑼加入了SVP,TZVP,QZV基组的密度拟合基。Fit关键字调用与AO基组匹配的拟合基,没有特定的拟合基时需要Auto关键字。
⑽在XXXXXXXXXXute文件中包含DensityFit关键字,只要执行纯密度泛函就使用拟合。
⑾为了与文献发表的基组兼容,读入的密度拟合因子可以是非归一化的原函数,密度归一化的原函数,或归一化的原函数。
========================================================================================================
以上来自锑度百科
我有Gaussian09W破解版+GaussView5.0破解版,如果在10天内需要的人数>10,我会发下载地址。。。
主要原因是Gaussian软件太为专业,于是需要的人数少得多。我的网盘地方有限,水管小,上传太慢。。。
[groupid=337]化学[/groupid]
Gaussian是一个功能强大的量子化学综合软件包。其可执行程序可在不同型号的大型计算机,超级计算机,工作站和个人计算机上运行,并相应有不同的版本。高斯功能:过渡态能量和结构、键和反应能量、分子轨道、原子电荷和电势、振动频率、红外和拉曼光谱、核磁性质、极化率和超极化率、热力学性质、反应路径,计算可以对体系的基态或激发态执行。可以预测周期体系的能量,结构和分子轨道。因此,Gaussian可以作为功能强大的工具,用于研究许多化学领域的课题,例如取代基的影响,化学反应机理,势能曲面和激发能等等。
Gaussian 09增加的功能
能量和求导
⑴最近发展的半经验模型(AM1,PM3,PM3MM,PDDG,PM6),计算解析一阶导和二阶导,用户自定义参数,以及结合使用PCM溶剂模型。
⑵TDDFT解析梯度和数值频率。
⑶EOM-CCSD计算激发能。
⑷新的DFT泛函,包括HSE,wB97,m05/m06,LC类泛函,以及双杂化B2PLYP。
⑸经验离散模型和相应的泛函。
⑹ROMP3,ROMP4,ROCCSD,ROCCSD(T)能量。
⑺W1RO,W1BD,G4方法计算能量。
⑻DFTB半经验模型,以及使用解析矩阵元的DFTBA版本。
ONIOM
⑴ONIOM与PCM组合。有多种ONIOM+PCM模型。
⑵ONIOM计算IRC,即使分子包含上千个原子效率也很高。
溶剂化
⑴新的PCM溶剂化算法,使能量成为核坐标的连续函数。现在,PCM的几何优化与气相优化的收敛速度一样。
⑵特定态的自洽溶剂化,用于模拟荧光和其它发射过程。它对上百种溶剂进行了参数化,可以给出非常好的总溶剂化自由能。
几何优化和IRC
⑴能量最小化默认使用GEDⅡS几何优化算法,这对大的柔软分子特别有帮助。
⑵对最小值和过渡结构使用二次收敛ONIOM(MO:MM)优化,既用于力学部分,也用于电子嵌入部分。
⑶一个输入部分,用于控制优化中的冻结或非冻结原子。可以用原子、元素、残基、或ONIOM层来指定原子。
分子特性
⑴解析的含频ROA强度。
⑵解析的DFT 超极化率。
⑶使用两个态的谐振模式,通过Franck-Condon原理计算电子激发、发射、光电离的谱带带型。
⑷用Herzberg-Teller或Franck-Condon-Herzberg-Teller理论计算电子激发的谱带带型。
⑸选择简正模式用于显示,非谐校正,和FC/HT/FCHT分析。可通过原子、元素、残基、或ONIOM层来选择。
分析和输出
⑴蛋白质二级结构的信息可以包含在分子指定输入部分,或者.fchk文件中。
⑵轨道布居分析,给出原子或角动量对轨道的贡献。
⑶正则UHF/UDFT进行二次正交化,用于显示或用于ROHF计算的初始猜测。
⑷CIS和TD激发的自然跃迁轨道分析。
⑸把占据轨道投影到最小基之后,进行Mulliken布居分析。当基组增大时,这能给出稳定的布居。
其它新功能
⑴SCF的初始猜测可以从片段计算的组合产生,需要指定每个片段的电荷和自旋。
⑵用四点差分而不是默认的两点差分计算数值频率,具有更高的精度和数值稳定性。
效率改善
⑴HF和DFT对大分子的频率计算更快,特别是当并行时。
⑵FMM以及线性标度的库仑和交换对簇并行。
⑶大体系的ONIOM(MO:MM)频率计算更快,特别是对电子嵌入。可以计算100-200 QM原子和6000 MM原子的频率。
⑷在大型频率计算中保存简正模式,用于显示或打印模式,以及开始IRC=RCFC任务。
⑸CC,BD,和EOM-CCSD振幅可以保存在检查点文件中,在以后的计算中读入。保存BD轨道并在以后读入。
⑹半经验,HF,和DFT的频率计算可以在中期计算中重新开始。
⑺CC和EOM-CC计算可以在中期计算中重新开始。
⑻ONIOM各步的初始猜测可以来自不同的检查点文件。
⑼加入了SVP,TZVP,QZV基组的密度拟合基。Fit关键字调用与AO基组匹配的拟合基,没有特定的拟合基时需要Auto关键字。
⑽在XXXXXXXXXXute文件中包含DensityFit关键字,只要执行纯密度泛函就使用拟合。
⑾为了与文献发表的基组兼容,读入的密度拟合因子可以是非归一化的原函数,密度归一化的原函数,或归一化的原函数。
========================================================================================================
以上来自锑度百科
我有Gaussian09W破解版+GaussView5.0破解版,如果在10天内需要的人数>10,我会发下载地址。。。
主要原因是Gaussian软件太为专业,于是需要的人数少得多。我的网盘地方有限,水管小,上传太慢。。。
[groupid=337]化学[/groupid]


引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
本帖最后由 718281828kc 于 2014-4-28 22:33 编辑
使用实例(这里用ChemBio3D 13.0作为输入和输出端)
通过安装特定格式的输入、输出文件,Gaussian可以与ChemBioOffice联用,也可直接用GaussView输入/输出,不用自己敲代码。。。
最终格式可以选择gif、jpg、tif、bmp等等普通图片格式和gjf——Gaussian专用格式、IS等等,同时显示计算报告。
Chem3D也可直接输出gjf格式,可与Gaussian进行hotlink连接。
最NB的是,输出格式gif可以为动画形式,甚至可以输出a♂v♂i。。。对着分子开*~~~
实例:DNDAF(二硝基偶氮氧化呋咱)和DNBF(二硝基联呋咱)的PM3水平结构优化
![Gaussian PM3.jpg]()
![DNDAF2.jpg]()
![DNDAF1.jpg]()
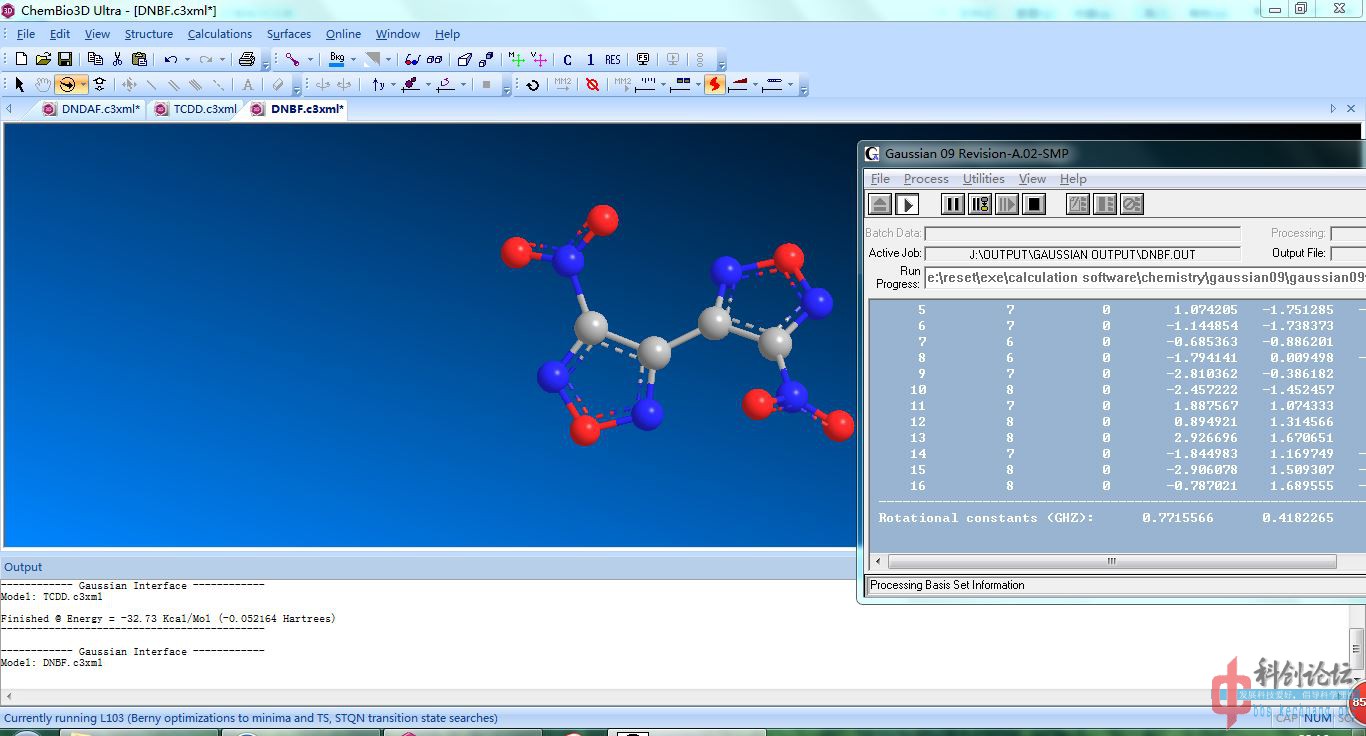
DNBF(计算中)
![Gaussian Minimize Calculation.jpg]()
使用实例(这里用ChemBio3D 13.0作为输入和输出端)
通过安装特定格式的输入、输出文件,Gaussian可以与ChemBioOffice联用,也可直接用GaussView输入/输出,不用自己敲代码。。。
最终格式可以选择gif、jpg、tif、bmp等等普通图片格式和gjf——Gaussian专用格式、IS等等,同时显示计算报告。
Chem3D也可直接输出gjf格式,可与Gaussian进行hotlink连接。
最NB的是,输出格式gif可以为动画形式,甚至可以输出a♂v♂i。。。对着分子开*~~~
实例:DNDAF(二硝基偶氮氧化呋咱)和DNBF(二硝基联呋咱)的PM3水平结构优化
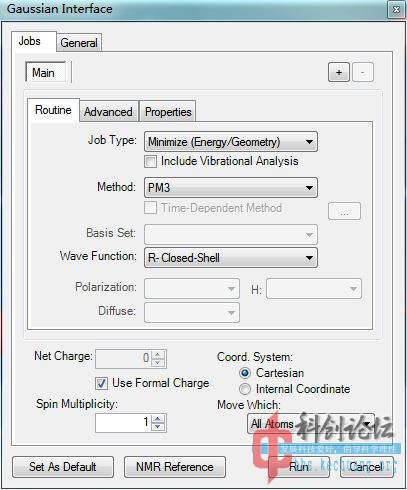
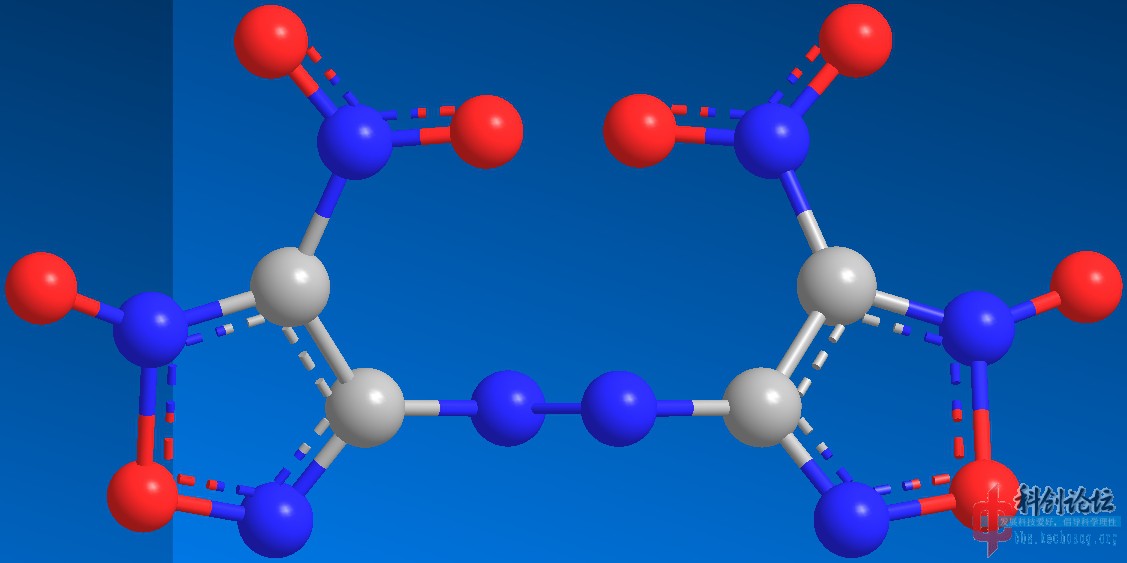
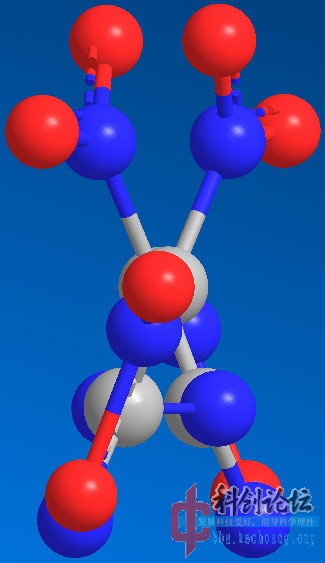
DNBF(计算中)
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
不要看那个A.02版本
我找到的是Gaussian09 B.01 for Windows,只是忘了升级
我找到的是Gaussian09 B.01 for Windows,只是忘了升级
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。


引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
魅影封喉 发表于 2014-4-28 22:39
233,最近就在汉化这个,工作量太大
是Gaussian03还是09?
A还是B。。。还是C?
我找不到windows系统的C版本,最高的仅为B.01。。。
最新的D.01只有Linux系统。。。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
718281828kc 发表于 2014-4-29 17:51
是Gaussian03还是09?
A还是B。。。还是C?
我找不到windows系统的C版本,最高的仅为B.01。。。
应该是03吧,我在破解这个的API,然后和我的软件对接的。
比如用B3LYP算个生成热,然后生成热数据可以直接进行爆炸参数估算
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
魅影封喉 发表于 2014-4-29 17:54
应该是03吧,我在破解这个的API,然后和我的软件对接的。
比如用B3LYP算个生成热,然后生成热数据可以直 ...
哦
03的bug比较多,算着算着就报错了
09A.01/02好得多,但还是有。B修正了大多数bug,包括A里的
听说C比B还难用。。。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
这是Gaussian09的中文版用户手册:

提示:如果是用于发表论文,请购买正版Gaussian程序或联系导师或与具有正版软件的科研部门合作,否则后果很严重。一旦被发现,就会面临终生禁用的处罚。
当然大多数人都是自己用233333
提示:如果是用于发表论文,请购买正版Gaussian程序或联系导师或与具有正版软件的科研部门合作,否则后果很严重。一旦被发现,就会面临终生禁用的处罚。
当然大多数人都是自己用233333
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
实例2:C4H4(正四面体烷)的DFT-B3LYP算法优化(原先的结构式是瞎连的)
![Gaussian DFT-B3LYP.jpg]()
部分输出结果
Input orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 0.307459 0.478730 -0.325944
2 6 0 -0.663338 -0.354605 0.454188
3 1 0 0.405403 1.272309 -1.034358
4 6 0 0.651798 0.030104 1.062187
5 6 0 0.606481 -0.965386 -0.056942
6 1 0 -1.697334 -0.533840 0.654040
7 1 0 1.049668 -1.852240 -0.454807
8 1 0 1.146163 0.301627 1.969436
---------------------------------------------------------------------
Distance matrix (angstroms):
1 2 3 4 5
1 C 0.000000
2 C 1.498499 0.000000
3 H 1.068276 2.450476 0.000000
4 C 1.498913 1.499082 2.449344 0.000000
5 C 1.499081 1.498915 2.450113 1.498500 0.000000
6 H 2.450481 1.068276 3.245669 2.450110 2.449345
7 H 2.449673 2.450219 3.242494 2.450059 1.068280
8 H 2.450216 2.449676 3.242488 1.068280 2.450059
6 7 8
6 H 0.000000
7 H 3.242490 0.000000
8 H 3.242491 3.244289 0.000000
Stoichiometry C4H4
Framework group C1[X(C4H4)]
Deg. of freedom 18
Full point group C1 NOp 1
Largest Abelian subgroup C1 NOp 1
Largest concise Abelian subgroup C1 NOp 1
Standard orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 -0.715880 0.218964 -0.531112
2 6 0 0.716347 -0.221740 -0.529329
3 1 0 -1.550819 0.473417 -1.147022
4 6 0 -0.220747 -0.714675 0.531857
5 6 0 0.220280 0.717451 0.528302
6 1 0 1.551834 -0.479420 -1.143151
7 1 0 0.475715 1.553663 1.142082
8 1 0 -0.476729 -1.547659 1.149783
---------------------------------------------------------------------
Rotational constants (GHZ): 13.4566117 13.4558540 13.4539132
**********************************************************************
Population analysis using the SCF density.
**********************************************************************
Orbital symmetries:
Occupied (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A)
(A) (A)
Virtual (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A)
(A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A)
(A) (A) (A) (A) (A) (A)
The electronic state is 1-A.
Alpha occ. eigenvalues -- -10.20120 -10.20081 -10.20079 -10.20076 -0.99224
Alpha occ. eigenvalues -- -0.58692 -0.58685 -0.58681 -0.52760 -0.39004
Alpha occ. eigenvalues -- -0.38965 -0.38956 -0.22555 -0.22526
Alpha virt. eigenvalues -- 0.12641 0.13552 0.13560 0.13583 0.15442
Alpha virt. eigenvalues -- 0.15447 0.15451 0.24556 0.24606 0.24616
Alpha virt. eigenvalues -- 0.50321 0.56313 0.56320 0.56330 0.56688
Alpha virt. eigenvalues -- 0.56699 0.69827 0.69829 0.69831 0.85266
Alpha virt. eigenvalues -- 0.85277 0.85284 0.87678 0.95137 0.95150
Alpha virt. eigenvalues -- 0.95172 1.50755 1.51018 1.51094 1.71072
Condensed to atoms (all electrons):
1 2 3 4 5 6
1 C 5.482948 0.098012 0.389280 0.098789 0.098916 -0.008737
2 C 0.098012 5.482948 -0.008737 0.098916 0.098791 0.389280
3 H 0.389280 -0.008737 0.496420 -0.008795 -0.008713 -0.000397
4 C 0.098789 0.098916 -0.008795 5.482959 0.098075 -0.008713
5 C 0.098916 0.098791 -0.008713 0.098075 5.482957 -0.008795
6 H -0.008737 0.389280 -0.000397 -0.008713 -0.008795 0.496420
7 H -0.008760 -0.008717 -0.000399 -0.008767 0.389285 -0.000399
8 H -0.008718 -0.008760 -0.000399 0.389284 -0.008767 -0.000399
7 8
1 C -0.008760 -0.008718
2 C -0.008717 -0.008760
3 H -0.000399 -0.000399
4 C -0.008767 0.389284
5 C 0.389285 -0.008767
6 H -0.000399 -0.000399
7 H 0.496418 -0.000399
8 H -0.000399 0.496419
Mulliken atomic charges:
1
1 C -0.141731
2 C -0.141733
3 H 0.141741
4 C -0.141748
5 C -0.141747
6 H 0.141741
7 H 0.141738
8 H 0.141738
Sum of Mulliken atomic charges = 0.00000
Mulliken charges with hydrogens summed into heavy atoms:
1
1 C 0.000010
2 C 0.000009
4 C -0.000010
5 C -0.000009
Sum of Mulliken charges with hydrogens summed into heavy atoms = 0.00000
Electronic spatial extent (au): <R**2>= 178.9393
Charge= 0.0000 electrons
Dipole moment (field-independent basis, Debye):
X= 0.0000 Y= 0.0000 Z= 0.0015 Tot= 0.0015
Quadrupole moment (field-independent basis, Debye-Ang):
XX= -22.5836 YY= -22.5889 ZZ= -22.6051
XY= -0.0016 XZ= 0.0000 YZ= 0.0000
Traceless Quadrupole moment (field-independent basis, Debye-Ang):
XX= 0.0089 YY= 0.0036 ZZ= -0.0126
XY= -0.0016 XZ= 0.0000 YZ= 0.0000
Octapole moment (field-independent basis, Debye-Ang**2):
XXX= 0.0050 YYY= 0.0292 ZZZ= 0.0099 XYY= 0.0115
XXY= -0.0120 XXZ= -3.7114 XZZ= -0.0165 YZZ= -0.0172
YYZ= 3.7116 XYZ= 2.5130
Hexadecapole moment (field-independent basis, Debye-Ang**3):
XXXX= -74.6539 YYYY= -74.6604 ZZZZ= -80.2382 XXXY= -3.7619
XXXZ= 0.0052 YYYX= 3.7517 YYYZ= 0.0293 ZZZX= 0.0072
ZZZY= -0.0423 XXYY= -26.8825 XXZZ= -21.3726 YYZZ= -21.3637
XXYZ= 0.0128 YYXZ= -0.0123 ZZXY= 0.0010
N-N= 1.046798475237D+02 E-N=-5.674416870799D+02 KE= 1.538193052986D+02
Test job not archived.
1|1|UNPC-MS-201201021153|FOpt|RB3LYP|6-31G|C4H4|ADMINISTRATOR|30-Apr-2
014|0||# RB3LYP/6-31G Opt Test||[No Title]||0,1|C,0.3074587049,0.47872
96157,-0.3259436192|C,-0.6633379245,-0.3546052984,0.454188444|H,0.4054
03297,1.2723091803,-1.0343584375|C,0.6517980058,0.030104343,1.06218688
16|C,0.6064808033,-0.9653855669,-0.0569420238|H,-1.6973342761,-0.53383
9574,0.6540396794|H,1.0496683567,-1.8522399297,-0.4548070772|H,1.14616
30329,0.3016272299,1.9694361527||Version=IA32W-G09RevB.01|State=1-A|HF
=-154.5741516|RMSD=1.955e-009|RMSF=7.364e-005|Dipole=0.0004491,-0.0002
938,0.0002447|Quadrupole=-0.0024785,0.0009637,0.0015148,0.0061681,-0.0
051901,0.0010897|PG=C01 [X(C4H4)]||@
PENOTUS DIED A HUNDRED YEARS OLD WANTING BUT TWO...
AND HE USED TO SAY BEFORE HE DIED, HAVING SPENT HIS
WHOLE LIFE IN VAINLY SEARCHING AFTER THE PHILOSOPHER'S
STONE, THAT IF HE HAD A MORTAL ENEMY HE DID NOT DARE
TO ENCOUNTER OPENLY, HE WOULD ADVISE HIM ABOVE ALL
THINGS TO GIVE HIMSELF UP TO THE STUDY AND PRACTICE
OF ALCHEMY. -- NICOLAS LEMERY, M.D.
"A COURSE OF CHYMISTRY", WALTER KETTILBY, LONDON, 1686
Job cpu time: 0 days 0 hours 2 minutes 33.0 seconds.
File lengths (MBytes): RWF= 5 Int= 0 D2E= 0 Chk= 1 Scr= 1
Normal termination of Gaussian 09 at Wed Apr 30 12:42:05 2014.

部分输出结果
Input orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 0.307459 0.478730 -0.325944
2 6 0 -0.663338 -0.354605 0.454188
3 1 0 0.405403 1.272309 -1.034358
4 6 0 0.651798 0.030104 1.062187
5 6 0 0.606481 -0.965386 -0.056942
6 1 0 -1.697334 -0.533840 0.654040
7 1 0 1.049668 -1.852240 -0.454807
8 1 0 1.146163 0.301627 1.969436
---------------------------------------------------------------------
Distance matrix (angstroms):
1 2 3 4 5
1 C 0.000000
2 C 1.498499 0.000000
3 H 1.068276 2.450476 0.000000
4 C 1.498913 1.499082 2.449344 0.000000
5 C 1.499081 1.498915 2.450113 1.498500 0.000000
6 H 2.450481 1.068276 3.245669 2.450110 2.449345
7 H 2.449673 2.450219 3.242494 2.450059 1.068280
8 H 2.450216 2.449676 3.242488 1.068280 2.450059
6 7 8
6 H 0.000000
7 H 3.242490 0.000000
8 H 3.242491 3.244289 0.000000
Stoichiometry C4H4
Framework group C1[X(C4H4)]
Deg. of freedom 18
Full point group C1 NOp 1
Largest Abelian subgroup C1 NOp 1
Largest concise Abelian subgroup C1 NOp 1
Standard orientation:
---------------------------------------------------------------------
Center Atomic Atomic Coordinates (Angstroms)
Number Number Type X Y Z
---------------------------------------------------------------------
1 6 0 -0.715880 0.218964 -0.531112
2 6 0 0.716347 -0.221740 -0.529329
3 1 0 -1.550819 0.473417 -1.147022
4 6 0 -0.220747 -0.714675 0.531857
5 6 0 0.220280 0.717451 0.528302
6 1 0 1.551834 -0.479420 -1.143151
7 1 0 0.475715 1.553663 1.142082
8 1 0 -0.476729 -1.547659 1.149783
---------------------------------------------------------------------
Rotational constants (GHZ): 13.4566117 13.4558540 13.4539132
**********************************************************************
Population analysis using the SCF density.
**********************************************************************
Orbital symmetries:
Occupied (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A)
(A) (A)
Virtual (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A)
(A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A) (A)
(A) (A) (A) (A) (A) (A)
The electronic state is 1-A.
Alpha occ. eigenvalues -- -10.20120 -10.20081 -10.20079 -10.20076 -0.99224
Alpha occ. eigenvalues -- -0.58692 -0.58685 -0.58681 -0.52760 -0.39004
Alpha occ. eigenvalues -- -0.38965 -0.38956 -0.22555 -0.22526
Alpha virt. eigenvalues -- 0.12641 0.13552 0.13560 0.13583 0.15442
Alpha virt. eigenvalues -- 0.15447 0.15451 0.24556 0.24606 0.24616
Alpha virt. eigenvalues -- 0.50321 0.56313 0.56320 0.56330 0.56688
Alpha virt. eigenvalues -- 0.56699 0.69827 0.69829 0.69831 0.85266
Alpha virt. eigenvalues -- 0.85277 0.85284 0.87678 0.95137 0.95150
Alpha virt. eigenvalues -- 0.95172 1.50755 1.51018 1.51094 1.71072
Condensed to atoms (all electrons):
1 2 3 4 5 6
1 C 5.482948 0.098012 0.389280 0.098789 0.098916 -0.008737
2 C 0.098012 5.482948 -0.008737 0.098916 0.098791 0.389280
3 H 0.389280 -0.008737 0.496420 -0.008795 -0.008713 -0.000397
4 C 0.098789 0.098916 -0.008795 5.482959 0.098075 -0.008713
5 C 0.098916 0.098791 -0.008713 0.098075 5.482957 -0.008795
6 H -0.008737 0.389280 -0.000397 -0.008713 -0.008795 0.496420
7 H -0.008760 -0.008717 -0.000399 -0.008767 0.389285 -0.000399
8 H -0.008718 -0.008760 -0.000399 0.389284 -0.008767 -0.000399
7 8
1 C -0.008760 -0.008718
2 C -0.008717 -0.008760
3 H -0.000399 -0.000399
4 C -0.008767 0.389284
5 C 0.389285 -0.008767
6 H -0.000399 -0.000399
7 H 0.496418 -0.000399
8 H -0.000399 0.496419
Mulliken atomic charges:
1
1 C -0.141731
2 C -0.141733
3 H 0.141741
4 C -0.141748
5 C -0.141747
6 H 0.141741
7 H 0.141738
8 H 0.141738
Sum of Mulliken atomic charges = 0.00000
Mulliken charges with hydrogens summed into heavy atoms:
1
1 C 0.000010
2 C 0.000009
4 C -0.000010
5 C -0.000009
Sum of Mulliken charges with hydrogens summed into heavy atoms = 0.00000
Electronic spatial extent (au): <R**2>= 178.9393
Charge= 0.0000 electrons
Dipole moment (field-independent basis, Debye):
X= 0.0000 Y= 0.0000 Z= 0.0015 Tot= 0.0015
Quadrupole moment (field-independent basis, Debye-Ang):
XX= -22.5836 YY= -22.5889 ZZ= -22.6051
XY= -0.0016 XZ= 0.0000 YZ= 0.0000
Traceless Quadrupole moment (field-independent basis, Debye-Ang):
XX= 0.0089 YY= 0.0036 ZZ= -0.0126
XY= -0.0016 XZ= 0.0000 YZ= 0.0000
Octapole moment (field-independent basis, Debye-Ang**2):
XXX= 0.0050 YYY= 0.0292 ZZZ= 0.0099 XYY= 0.0115
XXY= -0.0120 XXZ= -3.7114 XZZ= -0.0165 YZZ= -0.0172
YYZ= 3.7116 XYZ= 2.5130
Hexadecapole moment (field-independent basis, Debye-Ang**3):
XXXX= -74.6539 YYYY= -74.6604 ZZZZ= -80.2382 XXXY= -3.7619
XXXZ= 0.0052 YYYX= 3.7517 YYYZ= 0.0293 ZZZX= 0.0072
ZZZY= -0.0423 XXYY= -26.8825 XXZZ= -21.3726 YYZZ= -21.3637
XXYZ= 0.0128 YYXZ= -0.0123 ZZXY= 0.0010
N-N= 1.046798475237D+02 E-N=-5.674416870799D+02 KE= 1.538193052986D+02
Test job not archived.
1|1|UNPC-MS-201201021153|FOpt|RB3LYP|6-31G|C4H4|ADMINISTRATOR|30-Apr-2
014|0||# RB3LYP/6-31G Opt Test||[No Title]||0,1|C,0.3074587049,0.47872
96157,-0.3259436192|C,-0.6633379245,-0.3546052984,0.454188444|H,0.4054
03297,1.2723091803,-1.0343584375|C,0.6517980058,0.030104343,1.06218688
16|C,0.6064808033,-0.9653855669,-0.0569420238|H,-1.6973342761,-0.53383
9574,0.6540396794|H,1.0496683567,-1.8522399297,-0.4548070772|H,1.14616
30329,0.3016272299,1.9694361527||Version=IA32W-G09RevB.01|State=1-A|HF
=-154.5741516|RMSD=1.955e-009|RMSF=7.364e-005|Dipole=0.0004491,-0.0002
938,0.0002447|Quadrupole=-0.0024785,0.0009637,0.0015148,0.0061681,-0.0
051901,0.0010897|PG=C01 [X(C4H4)]||@
PENOTUS DIED A HUNDRED YEARS OLD WANTING BUT TWO...
AND HE USED TO SAY BEFORE HE DIED, HAVING SPENT HIS
WHOLE LIFE IN VAINLY SEARCHING AFTER THE PHILOSOPHER'S
STONE, THAT IF HE HAD A MORTAL ENEMY HE DID NOT DARE
TO ENCOUNTER OPENLY, HE WOULD ADVISE HIM ABOVE ALL
THINGS TO GIVE HIMSELF UP TO THE STUDY AND PRACTICE
OF ALCHEMY. -- NICOLAS LEMERY, M.D.
"A COURSE OF CHYMISTRY", WALTER KETTILBY, LONDON, 1686
Job cpu time: 0 days 0 hours 2 minutes 33.0 seconds.
File lengths (MBytes): RWF= 5 Int= 0 D2E= 0 Chk= 1 Scr= 1
Normal termination of Gaussian 09 at Wed Apr 30 12:42:05 2014.
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
justkaka 发表于 2014-5-3 11:01 有没有朋友知道免费的超算中心?
贵州大学有一个对外开放的量化计算网站,地址不记得了,自己搜索下。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
本帖最后由 718281828kc 于 2014-5-9 10:46 编辑
如果大家有需要。。。
那么还可以发MOPAC2012。。。
就4.2MB大小,是免费软件但不开源,可与Chem3D联用。
序列号可以免费获得。。。
MOPAC2012独家支持PM7算法,是半经验算法精度最高的,还可以解决例如-SO3Na之类使PM3报错的问题。
利用独家开发的MOZYME方法,可以在仅引入微量可忽略误差的情况下,大大加速运算。
还有DFT-D近似密度泛函方法,在保持高精确度的情况下,成数量级地加速运算。
例如
1. H2O Box,左图12426原子,右图25095原子,左图PM7 MOZYME 4线程 14.4min,-282616.10434kcal/mol,内存用了1.2GB;
右图PM7 MOZYME 4线程 98.6min,-576587.48001kcal/mol,内存用了4.5GB。
![H2O Box.jpg]()
2. ETK蛋白,3944原子
PM7 MOZYME 单线程 323.8/325.5s -16765.64836kcal/mol,用了300MB内存
![ETK Protein.png]()
如果大家有需要。。。
那么还可以发MOPAC2012。。。
就4.2MB大小,是免费软件但不开源,可与Chem3D联用。
序列号可以免费获得。。。
MOPAC2012独家支持PM7算法,是半经验算法精度最高的,还可以解决例如-SO3Na之类使PM3报错的问题。
利用独家开发的MOZYME方法,可以在仅引入微量可忽略误差的情况下,大大加速运算。
还有DFT-D近似密度泛函方法,在保持高精确度的情况下,成数量级地加速运算。
例如
1. H2O Box,左图12426原子,右图25095原子,左图PM7 MOZYME 4线程 14.4min,-282616.10434kcal/mol,内存用了1.2GB;
右图PM7 MOZYME 4线程 98.6min,-576587.48001kcal/mol,内存用了4.5GB。

2. ETK蛋白,3944原子
PM7 MOZYME 单线程 323.8/325.5s -16765.64836kcal/mol,用了300MB内存
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
简介
最著名的半经验分子轨道程序包。MOPAC从1981年由J. J. P. Stewart开始开发,最早基于Dewar和Thiel的NDDO方法。在发布了MOPAC 7.0之后,MOPAC成为Fujitsu公司的商业软件Fujitsu MOPAC。但是15年后,作者在MOPAC 7.0的基础上重新开始了非商业版本MOPAC的开发,用FORTRAN 90/95语言进行了改写,并加入了很多新的功能。MOPAC 2007是目前唯一由J. J. P. Stewart维护的MOPAC版本。MOPAC 2007(即MOPAC 7.2)功能相当于商业程序Fujitsu MOPAC 2002,增加了PM6,但缺少MINDO/3,PM5,解析导数,Tomasi溶剂模型和系间穿越。
功能
(7.0标准版)
1. MNDO,MINDO/3,AM1和PM3哈密顿量
2. 限制Hartree-Fock (RHF)和非限制Hartree-Fock (UHF)方法
3. 组态相互作用
(a) 100个组态
(b) 单重,双重,三重,四重,五重和六重
(c) 激发态
(d) 指定态的几何优化,等
4. 单自恰场计算
5. 几何优化
6. 梯度最小化
7. 过渡态定位
8. 反应路径坐标计算
9. 力常数计算
10. 简正坐标分析
11. 跃迁偶极计算
12. 热力学特性计算
13. 局域化轨道
14. 共价键顺序
15. 键分解为sigma和pi分量
16. 一维空间聚合物计算
17. 动力反应坐标计算
18. 内反应坐标计算
MOPAC 2007在MOPAC 7.0的基础上增加了以下功能:
1. 用FORTRAN 90改写。
2. 增强了周期体系(晶体、表面、聚合物)计算方面的功能。
3. 支持的方法有:MNDO,AM1,PM3,用于镧系元素的Sparkle/AM1,RM1,PM6,实现对所有主族元素及过渡金属的参数化。PM6是对NDDO方法的最近一次参数化,对近似做了三处修改,定义了对芯-芯相互作用的主要影响。除了12种镧系元素和所有的锕系元素之外,PM6参数已对大多数元素进行了优化。有机分子元素(H,C,N,O,F,P,S,Cl,Br,I)的参数已经稳定,其它主族元素、某些过渡元素以及1A和2A元素的参数尚在测试中。
4. 用PM6半经验模型模拟有机固体、无机固体的结构。
5. 用PM6计算氢键。
6. PM6对于仅由H,C,N,O,F,Si,P,S,Cl,Br和I构成的化合物计算极化率,平均误差为2.1%。
7. PM6更精确地计算生成热,熵,焓,热容,进行结构优化。对简单的有机分子精度最高,对复杂有机体系和无机体系精度稍低。PM6尚无法很好地计算频率。
8. 修改了AM1和PM3的一些严重错误。
9. 计算酸碱性(pKa)。
10.产生动画。
11.结构用JMol或CHIME显示。
MOPAC 2009的新功能:
1. 模拟酶的反应,优化整个蛋白质、DNA片断以及其它的大体系,可以进行直到15,000个原子的优化。
2. 线性标度算法MOZYME可以把半经验量子化学方法处理的极限从1,000个原子提高到15,000个(几何优化)或18,000个(SCF)原子,对100个原子以上的体系显著增加了计算速度。一个由1,000个原子构成的分子要比普通的MOPAC计算快60倍,并且只需要7%的内存。MOZYME可以与MOPAC的大多数功能结合使用,包括最新的PM6方法。
MOPAC 2012的新功能:
1. 加入新的半经验方法PM7和PM7-TS。
最著名的半经验分子轨道程序包。MOPAC从1981年由J. J. P. Stewart开始开发,最早基于Dewar和Thiel的NDDO方法。在发布了MOPAC 7.0之后,MOPAC成为Fujitsu公司的商业软件Fujitsu MOPAC。但是15年后,作者在MOPAC 7.0的基础上重新开始了非商业版本MOPAC的开发,用FORTRAN 90/95语言进行了改写,并加入了很多新的功能。MOPAC 2007是目前唯一由J. J. P. Stewart维护的MOPAC版本。MOPAC 2007(即MOPAC 7.2)功能相当于商业程序Fujitsu MOPAC 2002,增加了PM6,但缺少MINDO/3,PM5,解析导数,Tomasi溶剂模型和系间穿越。
功能
(7.0标准版)
1. MNDO,MINDO/3,AM1和PM3哈密顿量
2. 限制Hartree-Fock (RHF)和非限制Hartree-Fock (UHF)方法
3. 组态相互作用
(a) 100个组态
(b) 单重,双重,三重,四重,五重和六重
(c) 激发态
(d) 指定态的几何优化,等
4. 单自恰场计算
5. 几何优化
6. 梯度最小化
7. 过渡态定位
8. 反应路径坐标计算
9. 力常数计算
10. 简正坐标分析
11. 跃迁偶极计算
12. 热力学特性计算
13. 局域化轨道
14. 共价键顺序
15. 键分解为sigma和pi分量
16. 一维空间聚合物计算
17. 动力反应坐标计算
18. 内反应坐标计算
MOPAC 2007在MOPAC 7.0的基础上增加了以下功能:
1. 用FORTRAN 90改写。
2. 增强了周期体系(晶体、表面、聚合物)计算方面的功能。
3. 支持的方法有:MNDO,AM1,PM3,用于镧系元素的Sparkle/AM1,RM1,PM6,实现对所有主族元素及过渡金属的参数化。PM6是对NDDO方法的最近一次参数化,对近似做了三处修改,定义了对芯-芯相互作用的主要影响。除了12种镧系元素和所有的锕系元素之外,PM6参数已对大多数元素进行了优化。有机分子元素(H,C,N,O,F,P,S,Cl,Br,I)的参数已经稳定,其它主族元素、某些过渡元素以及1A和2A元素的参数尚在测试中。
4. 用PM6半经验模型模拟有机固体、无机固体的结构。
5. 用PM6计算氢键。
6. PM6对于仅由H,C,N,O,F,Si,P,S,Cl,Br和I构成的化合物计算极化率,平均误差为2.1%。
7. PM6更精确地计算生成热,熵,焓,热容,进行结构优化。对简单的有机分子精度最高,对复杂有机体系和无机体系精度稍低。PM6尚无法很好地计算频率。
8. 修改了AM1和PM3的一些严重错误。
9. 计算酸碱性(pKa)。
10.产生动画。
11.结构用JMol或CHIME显示。
MOPAC 2009的新功能:
1. 模拟酶的反应,优化整个蛋白质、DNA片断以及其它的大体系,可以进行直到15,000个原子的优化。
2. 线性标度算法MOZYME可以把半经验量子化学方法处理的极限从1,000个原子提高到15,000个(几何优化)或18,000个(SCF)原子,对100个原子以上的体系显著增加了计算速度。一个由1,000个原子构成的分子要比普通的MOPAC计算快60倍,并且只需要7%的内存。MOZYME可以与MOPAC的大多数功能结合使用,包括最新的PM6方法。
MOPAC 2012的新功能:
1. 加入新的半经验方法PM7和PM7-TS。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
就没有人再投票了吗?
就差1票了。。。
就差1票了。。。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。

在有机化学分子结构论文的时候,有的同学会用到这两个软件,搭配起来。挺强大的。
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
QQ578571918 发表于 2014-6-21 17:26
在有机化学分子结构论文的时候,有的同学会用到这两个软件,搭配起来。挺强大的。
想要的话可以看XXXXXXXXXXXXXXXXXXXXXXXX/t/65192
引用
加载评论中,请稍候...
200字以内,仅用于支线交流,主线讨论请采用回复功能。
引用
加载评论中,请稍候...

200字以内,仅用于支线交流,主线讨论请采用回复功能。